Human cells typically assemble a myriad of different proteins which constitute the work-force of the cellular environment and each of them is dedicated to a very specific function. The ability of a protein to perform its task is closely related to its structure: pockets, arms, and fingers are needed to store, move, and grasp molecules and other proteins resembling the workings of tiny biological machines.
All proteins are translated from messenger RNA into a sequence of amino acids. In order to perform their specific task, they have to fold and assemble into a specific three-dimensional structure. The instructions for proper folding are encoded in the amino acid sequence. Each amino acid carries different charged groups favouring certain interactions. Thus, a protein that contains errors in its sequence might struggle to fold properly. Since the charge of the amino acid is also related to its chemical microenvironment, folding might be compromised under oxidative or acidic conditions. Moreover, the folding process sometimes just fails, even when there are no sequence or environment changes.
Misfolded proteins expose portions of themselves that are normally buried on the inside, and thus interact with their environment in an abnormal way. Especially dangerous are exposed hydrophobic regions. They can compromise the cell compartmentalization by interacting with the membranes of organelles and vesicles; or they can stick together, forming compact fibrils that we call “amyloid”, due to their resemblance to starch (lat.: amylum). In these amyloid fibrils, the misfolded proteins interact non-specifically with each other using their amino acid backbone, rather than the residues that are normally exposed to the surface. This results in the formation of extensive β-sheets, held together by a large number of hydrogen bonds, that are very stable and almost impossible to break down. Moreover, amyloid fibrils seem to be able to trap other proteins and seed their conversion into more amyloid structures i.e. spreading. Even though it entails loss of function, aggregation is thought to have a protective effect: the more misfolded proteins interact with each other and aggregate, the less they are available for interaction with membranes or available for spreading.
Protein misfolding manifests itself in a plethora of diseases. They are collectively called amyloidoses, since they are associated with the deposition of amyloids in form of extracellular plaques or intracellular inclusions which may present systemically, or localised. The type and localisation of aggregates can be traced back to the protein that is affected by misfolding. It is still unclear, however, if the aggregates themself play a role in the pathology, or if they are symptomatic of an underlying issue.
The most well-studied amyloid diseases are the neurodegenerative diseases Alzheimer’s disease (AD) and Parkinson’s disease (PD). In AD, deposition of amyloids in form of extracellular plaques as well as intracellular neurofibrillary tangles is observed. The extracellular deposits can be led back to abnormal proteolytic cleavage of the Amyloid Precursor Protein (APP) which produces amyloidogenic Aβ peptides which form extracellular deposits in close proximity to the synaptic cleft. Intracellular deposits observed in patients with AD originate from a hyperphosphorylated form of tau protein. Soluble tau protein normally contributes in regulating microtubule assembly. Under pathological conditions, it becomes hyperphosphorylated and prone to aggregation forming insoluble filaments and tangled clumps, known as neurofibrillary tangles (NFTs). NFTs accumulate inside the neuronal cell body and survive even after the death of affected neuronal cells, where they may be released extracellularly. In PD, amyloid aggregates are mostly intracellular and appear as spherical masses in the cytoplasm. These have first been described by Fritz Heinrich Lewy in 1910 and are since called Lewy bodies. Their major component is α-synuclein. In physiological conditions, α-synuclein regulates synaptic vesicle-release and possibly cytoskeletal assembly. However, this small pre-synaptic protein is characterised by an intrinsically disordered structure that makes it prone to aggregation. When aggregated, α-synuclein can interact with membranes and, for instance, make neurotransmitter vesicles “leaky”. This is especially important in dopaminergic neurons, as the interaction between dopamine and α-synuclein seems to facilitate further α-synuclein oligomerization. This might be the reason why dopaminergic neurons of the substantia nigra in the midbrain are the first ones to degenerate in the patients. This particular region of the brain has a role in suppressing involuntary movements, and its loss is responsible for the characteristic tremors.
Many more forms of amyloid diseases exist and to date, 37 human proteins have been found to form amyloid deposits in pathology. While it is under debate, if the amyloid aggregates cause or contribute to the pathology, they are an important hallmark for the diagnosis of underlying disease. Due to their involvement in debilitating and progressive diseases, amyloids are associated with a negative connotation in general. However, a plethora of functional amyloids exists in humans and other organisms.
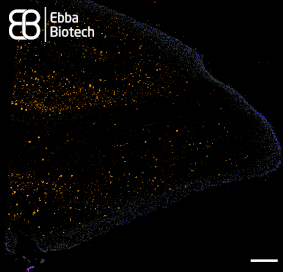
With our family of Amytracker molecules, Ebba Biotech provides a toolbox for sensitive, fast and safe detection of functional and disease-associated amyloid aggregates (See image showing a temporal lobe tissue section with pronounced AD pathology labelled with Amtracker 680 and DAPI counterstain. Tiled image at 10X magnification (Ex: 405 nm, Em1: 415-456 nm, Ex2: 587-694 nm), scale bar: 1mm).
Read More:
- Dobson, C.M. Nature (2003) Protein folding and misfolding. 426, 884–890
- Mohandas, E. et al. (2009) Neurobiology of Alzheimer's disease. Indian J Psychiatry 51(1), 55-61
- Kametani, F. and Hasegawa, M. (2018) Reconsideration of Amyloid Hypothesis and Tau Hypothesis in Alzheimer's Disease. Front Neurosci 12(25)
- Kumar, K. et al. (2018) Recent advances in the neurobiology and neuropharmacology of Alzheimer's disease. Biomed Pharmacother 98, 297-307